For long time, i know the Hartree-Fock method is a way to find the mean-field, and the method is a mean-field theory. But how exactly the Hartree-Fock connects to the mean field, I have no idea. And occasionally, I mentioned the Hartree-Fock in its simplest form. Here, I will makes the connection crystal clear.
Hartree = self-consistence field
Fock = anti-symmetry wave function
Mean-field approximation, or in chemistry, self-consistence field approximation.
The full Hamiltonian is



Here,  is one-body operator, and
is one-body operator, and  is two-body operator. I used Coulomb potential in the one-body operator, but it can be generalized as
is two-body operator. I used Coulomb potential in the one-body operator, but it can be generalized as  , and the mutual interaction can also be generalized as
, and the mutual interaction can also be generalized as  .
.
The idea of the mean-field approximation is that, what if, we can find a one-body potential  , the mean-field, such that
, the mean-field, such that



Here,  is the mean-field Hamiltonian, which represent most of the effective interaction to a particle, such that
is the mean-field Hamiltonian, which represent most of the effective interaction to a particle, such that  is the residual interaction, which is very small, and can be later treated as perturbation.
is the residual interaction, which is very small, and can be later treated as perturbation.
So, the problem is, how to find this mean field  ?
?
Back to the form  , we first construct a trial wave function,
, we first construct a trial wave function,



Here,  is the permutation operator, it can be 1-body exchange, 2-body exchange, and so on, but we will see that, only 1-body exchange (which is no change at all) and 2-body exchange are needed.
is the permutation operator, it can be 1-body exchange, 2-body exchange, and so on, but we will see that, only 1-body exchange (which is no change at all) and 2-body exchange are needed.  is the Hartree wave function, it is a simple product of wave function of difference particles of difference states, or the diagonal product of the Slater determinant. In $\phi_\lambda(i) $, $\latex \lambda$ represents the state and
is the Hartree wave function, it is a simple product of wave function of difference particles of difference states, or the diagonal product of the Slater determinant. In $\phi_\lambda(i) $, $\latex \lambda$ represents the state and  is the “id” of the particle. Notice that
is the “id” of the particle. Notice that

 is the anti-symmetrization operator, it commute with Hamiltonian and a kind of projector operator,
is the anti-symmetrization operator, it commute with Hamiltonian and a kind of projector operator,
![[A,H] = 0, A^2 = A.](https://s0.wp.com/latex.php?latex=%5BA%2CH%5D+%3D+0%2C+%C2%A0A%5E2+%3D+A.&bg=fafad3&fg=6f5e4e&s=0&c=20201002)
Now, we evaluate the energy using this trial wave function.



Since the one-body operator  only acts on the particle, any exchange will make the operator do nothing on the
only acts on the particle, any exchange will make the operator do nothing on the  particle, then the orthogonality of the wave function makes the integration zero. Lets demonstrate on 2 particles case.
particle, then the orthogonality of the wave function makes the integration zero. Lets demonstrate on 2 particles case.

Notice that this is the one-body operator act on particle 1.
OK,



The  is direct term, and the
is direct term, and the  is the exchange term. This is a simplified notation, the first position is for the -th particle, and the second position is always for the -th particle.
is the exchange term. This is a simplified notation, the first position is for the -th particle, and the second position is always for the -th particle.
Thus, the total energy is

We can factor out the ket  in the above equation and get the Fock-operator, with a notation for the exchange term operator
in the above equation and get the Fock-operator, with a notation for the exchange term operator



Becareful on the  ! I know the notation is messy, I know….
! I know the notation is messy, I know….
The Fock-operator is an effective one-body operator.
First, we put the trial basis wave function  can get the Fock-matrix
can get the Fock-matrix  , then diagonalize it, get the new eigen-states. Use this set of new eigen-states to calculate agian and again until converged!
, then diagonalize it, get the new eigen-states. Use this set of new eigen-states to calculate agian and again until converged!
So, where is the mean-field? Lets expand the Fock-operator into the Hartree-Fock equation.



This is the mean-field!
Using the trial basis, we evaluate the Fock-matrix, that is equivalent to evaluate the mean-field.

In this post, the Hartree-fock for 2-body ground state is discussed. Unfortunately, that method is not the same in here. I would said, that method is only Hartree but not Fock. Since the method in that post can find a consistence field, but the ground state spatial is not anti-symmetric.
Since the spin-state is factored out, the spatial wave function of the case is identical. Thus, the exchange term is gone. The Slater determinant is

Here,  are spin-state.
are spin-state.
In general, the 2 particle system, the energy is


When  , the direct term and exchange term cancelled. Thus, the “mean-field” is simply the Coulomb potential. Therefore, the method in that post is kind of getting around.
, the direct term and exchange term cancelled. Thus, the “mean-field” is simply the Coulomb potential. Therefore, the method in that post is kind of getting around.
Some people may found that the Fock operator is

In this way, the direct term for  will not be cancelled. In the case of 2 particle, the mean field is the Coulomb potential plus the average mutual interaction from the other particle.
will not be cancelled. In the case of 2 particle, the mean field is the Coulomb potential plus the average mutual interaction from the other particle.









 out.
out.








 . And the energy is in unit of Hartree,
. And the energy is in unit of Hartree,  .
.
 , because in the integration
, because in the integration  , the
, the  can be cancelled. Also, the
can be cancelled. Also, the  is a compact index of the orbital.
is a compact index of the orbital.



 .
.
 .
.
 from the Clebsch-Gordon coefficient, only a few terms need to be calculated.
from the Clebsch-Gordon coefficient, only a few terms need to be calculated.






 . Thus, if we flatten the matrix of matrixes, it is Hermitian, or symmetric.
. Thus, if we flatten the matrix of matrixes, it is Hermitian, or symmetric. is a matrix of matrixes. The inner matrix runs for
is a matrix of matrixes. The inner matrix runs for  indexes, while the bigger matrix runs for
indexes, while the bigger matrix runs for  index. This will simplify our minds.
index. This will simplify our minds.

 runs from 1 to 2. And remember the
runs from 1 to 2. And remember the  is a matrix of matrixes, using the identify
is a matrix of matrixes, using the identify  the sum becomes:
the sum becomes: ,
, represent the bigger matrix in
represent the bigger matrix in 







 .
.





 energy is under estimated.
energy is under estimated. , we can see, the mutual interaction between 1s and 2s state is attractive. While the interaction between 1s-1s and 2s-2s states are repulsive. The repulsive can be easily understood. But I am not sure how to explain the attractive between 1s-2s state.
, we can see, the mutual interaction between 1s and 2s state is attractive. While the interaction between 1s-1s and 2s-2s states are repulsive. The repulsive can be easily understood. But I am not sure how to explain the attractive between 1s-2s state. , so the missing 0.2 eV should be due to something else, for example, the incomplete basis set.
, so the missing 0.2 eV should be due to something else, for example, the incomplete basis set.
 . If 4s orbital included, the
. If 4s orbital included, the  . We can expect, if more orbital in included, the
. We can expect, if more orbital in included, the  will approach to
will approach to  .
.
 , the consequence is no mean field for the same state. Suppose the mean field energy is constructed using the ground state, and we only use 3 states, the direct term is
, the consequence is no mean field for the same state. Suppose the mean field energy is constructed using the ground state, and we only use 3 states, the direct term is






 , the direct term and the exchange term are identical, and then the mean field energy is zero. Also, when
, the direct term and the exchange term are identical, and then the mean field energy is zero. Also, when  the mean field energy is also zero.
the mean field energy is also zero. with
with  or
or 





 ,
,  ,
, 

 is,
is,
 , but the exchange term is multiply with
, but the exchange term is multiply with  , which are two different functions. i.e., the “mean field” is affecting two functions, or the “mean field” is shared by two states.
, which are two different functions. i.e., the “mean field” is affecting two functions, or the “mean field” is shared by two states.

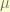


 , we set the first trial of the solution to be
, we set the first trial of the solution to be

 is used for the particle-j.
is used for the particle-j. , then we construct a new trial function and iterate until converge.
, then we construct a new trial function and iterate until converge.
 ,
,  is number state to use.
is number state to use.

 potential to be the basis,
potential to be the basis,

 . We assume,
. We assume,




 , because mixing parity will result non-reasonable result. ( why ? )
, because mixing parity will result non-reasonable result. ( why ? ) , and solve for the eigen value and eigen vector,
, and solve for the eigen value and eigen vector,


 for simplicity, the Hartree ground state is 1.7125 for both basis. The eigen wavefunction for the 2-basis is
for simplicity, the Hartree ground state is 1.7125 for both basis. The eigen wavefunction for the 2-basis is


 (orange) and
(orange) and  (blue). We can see that the wave function becomes little spread out.
(blue). We can see that the wave function becomes little spread out.





 (orange) and $ E \Psi^2 $ (blue), and there difference is the following
(orange) and $ E \Psi^2 $ (blue), and there difference is the following

 has to sum over all other particle. When all particles are in the same states, the sum is simple. For particles distribute among different states, in general, the sum is
has to sum over all other particle. When all particles are in the same states, the sum is simple. For particles distribute among different states, in general, the sum is
 , which is in state
, which is in state 










 contains 2 variables.
contains 2 variables.


 , so that,
, so that,
 is the single particle energy, and
is the single particle energy, and  is the single particle wave function.
is the single particle wave function. , construct a first trial total wavefuction
, construct a first trial total wavefuction 









 and
and  , we get the Hartree-Fock single particle Hamiltonian,
, we get the Hartree-Fock single particle Hamiltonian,




