In the past, we solved the radial wavefunction for the Woods-Saxon potential by Runge-Kutta method. But there is another method based on linear algebra, that all wave function can be expanded as a linear combination of a set of basis. In here, we use a free wave as basis, which is the spherical Bessel function.
to be annoying due to my poor memory , lets state the Schrödinger equation again,

Separating the radial part

replace



simplify

We can define a “free wave” Hamiltonian as

In this post, we explored the orthogonality of the the Riccati-Bessel function, and it is the eigen state for 



Thus, the Riccati-Bessel function can be served as the set of basis functions that span the whole space.
The total Hamiltonian is then

Since the solution can be expressed as a linear combination of the basis functions, then


Multiple 

The above is a matrix equation

Everything seems OK. But when I test the theory using Mathematica, it fail. For simplicity, we use the s-orbital, and the basis is

The Woods-Saxon parameters are 
Using Runge-Kutta method, the ground state energy is -36.09 MeV, and the wave function is

We set 


where 
After calculate the matrix, find the eigenvalue and eigenstate. The lowest eigen energy is -465.18 MeV, which indicate something wrong. And the wave function, constructed by the eigenstate look likes

The shape of the wavefunction looks similar to the correct wavefunction, but narrower and larger amplitude.
The next eigen wave function also look like 1s-orbital. The spectrum of the eigenvalues, which are the eigen energies, has 7 bound states.
I don’t know the problems….
One possible reason could be that the basis function is always 


I use the same basis and extract the coefficient,

And it can reproduce the original wave function at

Since the basis function is a sine wave and we only sum limited term, the long range oscillation can not be suppressed.
Another possible reason is that the basis is not normalizable. This root at the orthogonality of Bessel function that

An-another possible reason is that, the basis is for unbound state. But in this post, we used a not-normalizable, unbounded basis to construct the bound state wave function. It can form the solution correctly at short-range, but it is periodic. This is the nature of series expansion, always periodic.
In this sense, we are also expanding the solution into a series, that the series only produce periodic function, therefore, a correct way to do is using the spherical Hankel transform, which is transform the Schrodinger equation into momentum space.


 potential to be the basis,
potential to be the basis,

 . We assume,
. We assume,




 , because mixing parity will result non-reasonable result. ( why ? )
, because mixing parity will result non-reasonable result. ( why ? ) , and solve for the eigen value and eigen vector,
, and solve for the eigen value and eigen vector,


 for simplicity, the Hartree ground state is 1.7125 for both basis. The eigen wavefunction for the 2-basis is
for simplicity, the Hartree ground state is 1.7125 for both basis. The eigen wavefunction for the 2-basis is


 (orange) and
(orange) and  (blue). We can see that the wave function becomes little spread out.
(blue). We can see that the wave function becomes little spread out.





 (orange) and $ E \Psi^2 $ (blue), and there difference is the following
(orange) and $ E \Psi^2 $ (blue), and there difference is the following

 has to sum over all other particle. When all particles are in the same states, the sum is simple. For particles distribute among different states, in general, the sum is
has to sum over all other particle. When all particles are in the same states, the sum is simple. For particles distribute among different states, in general, the sum is
 , which is in state
, which is in state  . For example, if the system consists of 2 particles on state-1, and 2 particles on state-2. The mean field for the particle in state-1 is constructed using 1 particles on state-1 and 2 particles on state-2. Thus the energy for the state-2 may be not correct.
. For example, if the system consists of 2 particles on state-1, and 2 particles on state-2. The mean field for the particle in state-1 is constructed using 1 particles on state-1 and 2 particles on state-2. Thus the energy for the state-2 may be not correct.
 . Some text book
. Some text book  because the number of node is
because the number of node is  .
.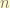 and
and  can take any integers.
can take any integers.  is
is







 is Bohr radius,
is Bohr radius,



 ,
,  .
.

 . We can see, when
. We can see, when  , the minimize point of the potential is at
, the minimize point of the potential is at  . The potential is vary shallow, and the bound state is very diffuses. Since the 2nd derivative of the wave function
. The potential is vary shallow, and the bound state is very diffuses. Since the 2nd derivative of the wave function  around the minimum point is
around the minimum point is
 ,
,  is
is is
is  .
. , the potential is also very shallow. For such a picture, we can see the wave function must be oscillating that the number of node must be larger then 1, so the principle quantum number should be larger than 1.
, the potential is also very shallow. For such a picture, we can see the wave function must be oscillating that the number of node must be larger then 1, so the principle quantum number should be larger than 1.
![a = 0.6 [fm]](https://s0.wp.com/latex.php?latex=a+%3D+0.6+%5Bfm%5D&bg=fafad3&fg=6f5e4e&s=0&c=20201002) . For simplicity, we use
. For simplicity, we use

 there is a “well” that can bound a nucleon.
there is a “well” that can bound a nucleon.


 at ground state.
at ground state.


 ” of Laplace equation
” of Laplace equation



 are always larger or equal the minimum energy.
are always larger or equal the minimum energy.








 is the interaction between the 2 particles.
is the interaction between the 2 particles.

 or
or  , and integrate
, and integrate  , we have
, we have


 is the mutual interaction, and
is the mutual interaction, and  are independent.
are independent.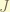 is the energy from the interaction of the particles.
is the energy from the interaction of the particles. is the energy due to EXCHANGE of the particles.
is the energy due to EXCHANGE of the particles. .
. in
in  can be reduced to
can be reduced to  are in difference orbits. When
are in difference orbits. When  ,
,  .
.


 is the shift of energy due to the mutual interaction.
is the shift of energy due to the mutual interaction. .
.










 contains 2 variables.
contains 2 variables.













 has to be 0. (home work)
has to be 0. (home work)














