After long preparation, I am ready to do this problem.
The two electron in the helium ground state occupy same spacial orbital but difference spin. Thus, the total wavefunction is

Since the Coulomb potential is spin-independent, the Hartree-Fock method reduce to Hartree method. The Hartree operator is

where the single-particle Hamiltonian and mutual interaction are


In the last step, we use atomic unit, such that 

We are going to use Hydrogen-like orbital as a basis set.

I like the left the 



Using basis set expansion, we need to calculate the matrix elements of


Now, we will concentrate on evaluate the mutual interaction integral.
Using the well-known expansion,

The angular integral

where the integral 
From this post, the triplet integral of spherical harmonic is easy to compute.

The Clebsch-Gordon coefficient imposed a restriction on 
The radial part,

The algebraic calculation of the integral is complicated, but after the restriction of 
The general consideration is done. now, we use the first 2 even states as a basis set.


These are both s-state orbital. Thus, the Clebsch-Gordon coefficient

The radial sum only has 1 term. And the mutual interaction becomes

The angular part

Thus, the mutual interaction energy is

The radial part

We can easy to see that 
We also notice that 


Now, we can start doing the Hartree method.
The general solution of the wave function is

The Hartree matrix is

In this simple 2-states example, index 



where the 
The first trial wave function is the Hydrogen-like orbital,


Solve for eigen system, we have the energy after 1st trial,

After 13th trial,


Thus, the mixing of the 2s state is only 3.7%.
Since the eigen energy contains the 1-body energy and 2-body energy. So, the total energy for 2 electrons is

In which ,

So the energies for
From He to He++.
From He+ to He++, 
From He to He+, is


The experimental 1 electron ionization energy for Helium atom is



The difference with experimental value is 2.175 eV. The following plot shows the Coulomb potential, the screening due to the existence of the other electron, the resultant mean field, the energy, and


Usually, the Hartree method will under estimate the energy, because it neglected the correlation, for example, pairing and spin dependence. In our calculation, the 
From the 
Since the mass correction and the fine structure correction is in order of 
If the basis set only contain the 1s orbit, the mutual interaction is 1.25 Hartree = 34.014 eV. Thus, the mixing reduce the interaction by 5.07 eV, just for 3.7% mixing
I included the 3s state,

The mutual energy is further reduced to 1.05415 Hartree = 28.6848 eV. The 




 , the consequence is no mean field for the same state. Suppose the mean field energy is constructed using the ground state, and we only use 3 states, the direct term is
, the consequence is no mean field for the same state. Suppose the mean field energy is constructed using the ground state, and we only use 3 states, the direct term is






 , the direct term and the exchange term are identical, and then the mean field energy is zero. Also, when
, the direct term and the exchange term are identical, and then the mean field energy is zero. Also, when  the mean field energy is also zero.
the mean field energy is also zero. with
with  or
or 





 ,
,  ,
, 

 is,
is,
 , but the exchange term is multiply with
, but the exchange term is multiply with  , which are two different functions. i.e., the “mean field” is affecting two functions, or the “mean field” is shared by two states.
, which are two different functions. i.e., the “mean field” is affecting two functions, or the “mean field” is shared by two states.







 , which is the eigen vector. Then, new trial functions with the new coefficients are used.
, which is the eigen vector. Then, new trial functions with the new coefficients are used.



 -particle in state
-particle in state 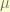


 , we set the first trial of the solution to be
, we set the first trial of the solution to be

 is used for the particle-j.
is used for the particle-j. , then we construct a new trial function and iterate until converge.
, then we construct a new trial function and iterate until converge.
 ,
,  is number state to use.
is number state to use.

 potential to be the basis,
potential to be the basis,

 . We assume,
. We assume,




 , because mixing parity will result non-reasonable result. ( why ? )
, because mixing parity will result non-reasonable result. ( why ? ) , and solve for the eigen value and eigen vector,
, and solve for the eigen value and eigen vector,


 for simplicity, the Hartree ground state is 1.7125 for both basis. The eigen wavefunction for the 2-basis is
for simplicity, the Hartree ground state is 1.7125 for both basis. The eigen wavefunction for the 2-basis is


 (orange) and
(orange) and  (blue). We can see that the wave function becomes little spread out.
(blue). We can see that the wave function becomes little spread out.





 (orange) and $ E \Psi^2 $ (blue), and there difference is the following
(orange) and $ E \Psi^2 $ (blue), and there difference is the following

 has to sum over all other particle. When all particles are in the same states, the sum is simple. For particles distribute among different states, in general, the sum is
has to sum over all other particle. When all particles are in the same states, the sum is simple. For particles distribute among different states, in general, the sum is
 , which is in state
, which is in state 





 . The exact solution is
. The exact solution is  .
. controls the wavelength of the plane wave. By increasing the number of wave
controls the wavelength of the plane wave. By increasing the number of wave 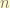 , we effectively control the minimum wavelength
, we effectively control the minimum wavelength  Therefore, larger the
Therefore, larger the 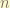 , the energy will approach to the actual value.
, the energy will approach to the actual value. , we need more number of wave to go to the actual energy. When small
, we need more number of wave to go to the actual energy. When small  , the energy is
, the energy is  , the difference is
, the difference is  or 0.02% difference.
or 0.02% difference.




 and
and  .
.




 are always larger or equal the minimum energy.
are always larger or equal the minimum energy.








 still in the space,
still in the space,
 , the rotation can be done by a rotational matrix,
, the rotation can be done by a rotational matrix,
 also from a basis. And the rotation on this new basis was
also from a basis. And the rotation on this new basis was 
 . The space becomes “3-dimensional” because
. The space becomes “3-dimensional” because  , otherwise, it will becomes “4-dimensional”.
, otherwise, it will becomes “4-dimensional”. , and further decomposed into
, and further decomposed into  .
. , the rotation of the function is simply multiplied by the rotation matrix.
, the rotation of the function is simply multiplied by the rotation matrix.
 is
is
 indicates that a rotation does not mix the spaces. The transformation of the eigen function is
indicates that a rotation does not mix the spaces. The transformation of the eigen function is


 .
.

 serves as the basis for eigenvalue of
serves as the basis for eigenvalue of  , eigen spaces for difference
, eigen spaces for difference  .
.
 , which is the primary base that contains only
, which is the primary base that contains only  .
. , the space has
, the space has  is unchanged after rotation. Thus, the size of the space reduced
is unchanged after rotation. Thus, the size of the space reduced  .
. , the space has
, the space has  , this time, the linear combinations
, this time, the linear combinations  behave like
behave like  and
and  behave like
behave like  , thus the size of the space reduce to
, thus the size of the space reduce to  .
. is
is  , and we can find
, and we can find  repeated combinations, thus the size of the
repeated combinations, thus the size of the  is
is  . We can find
. We can find  repeated combination, thus, the size of the irreducible space of order
repeated combination, thus, the size of the irreducible space of order  .
.





 will produce a coefficient related to the order, and the radial part can be extracted.
will produce a coefficient related to the order, and the radial part can be extracted.

 , where
, where  is the reduced Planck constant, which has the dimension of angular momentum.
is the reduced Planck constant, which has the dimension of angular momentum.
 . The
. The 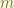 is the degeneracy for same eigenvalue
is the degeneracy for same eigenvalue 
 , such that
, such that

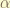 is the id of the basis. One can use
is the id of the basis. One can use 
 . Suppose the eigen vector is
. Suppose the eigen vector is

 on the left, but in general, the basis set are not orthogonal.
on the left, but in general, the basis set are not orthogonal.

 ,
,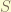 is
is
 is a diagonal matrix of eigen value. Now, we define a new non-unitary matrix
is a diagonal matrix of eigen value. Now, we define a new non-unitary matrix


 is a transform that from one basis to another basis, i.e.
is a transform that from one basis to another basis, i.e.





 , get the eigen system, then use
, get the eigen system, then use 




 is
is





![H_S = [H_S]_{ij} = \phi_i \cdot H\cdot \phi_j = \begin{pmatrix} 2 & 2 \\ 2 & 3 \end{pmatrix}](https://s0.wp.com/latex.php?latex=H_S+%3D+%5BH_S%5D_%7Bij%7D+%3D+%5Cphi_i+%5Ccdot+H%5Ccdot+%5Cphi_j+%3D+%C2%A0%5Cbegin%7Bpmatrix%7D+2+%26+2+%5C%5C+2+%26+3+%5Cend%7Bpmatrix%7D%C2%A0&bg=fafad3&fg=6f5e4e&s=0&c=20201002)







